
Integrative omics to study gene expression regulation
Gene expression is regulated in part by the incorporation of histone variants and by the dynamic modification of histones by various chemical groups. A tight regulation of these mechanisms is necessary during cellular differentiation processes; dysregulations are at stake in numerous diseases, including developmental defects and cancers.
The identity of histone sequence variants, which may differ by just a handful of amino acids over the protein sequence, can be precisely established by the proteomic analysis of histones purified from cells or tissues of interest. To confidently carry out such analyses, we established a database containing an exhaustive and non-redundant list of histone sequences for mouse and human (El Kennani et al., Epigenetics and Chromatin 2017). In the context of mouse spermatogenesis that is characterized by very dynamic gene expression changes and dramatic chromatin remodeling, we demonstrated the feasibility of identifying and quantifying numerous variants of histones H2A and H2B, including sequences that differ by a single amino acid (El Kennani et al., Epigenetics and Chromatin 2018).
Our research activities also aim at studying the effects on gene expression of recently described histone post-translational modifications (PTMs) that target lysine residues: those resemble acetylation, yet differ in length and hydrophobicity, and are collectively called acylations. We started such explorations in the context of mouse spermatogenesis (collaboration Julie Cocquet, Institut Cochin, Paris), a differentiation process in which lysine crotonylation was originally reported (Tan M. et al., Cell 2011). Quite unexpectedly, the proteomic analysis of histones extracted from three cellular stages of mouse spermatogenesis revealed that crotonylation was of similar stoichiometry to acetylation on histone H3 Lysine 27 (H3K27). Since H3K27ac marks active transcription when present at promoters, and also marks active distal enhancers, we combined our proteomics data with ChIP-seq data obtained on H3K27cr to get its localization on the genome, and to public RNA-seq data to study the effect of this histone PTM on gene expression (collaboration with Christophe Battail, CEA Grenoble). Additional public ChIP-seq datasets were collected to compare the association of various chromatin-binding proteins with H3K27cr and with H3K27ac. Metabolomics data on the donor groups for acylations, namely acyl-CoA, were also acquired (collaboration with LEMM, CEA Saclay), to try to correlate the abundance of these analytes with histone PTM stoichiometries. Finally, our study indicated both synergistic and specific actions of each histone modification at promoters and distal enhancers (Crespo M. et al. Nucleic Acids Res. 2020)
Some granted projects
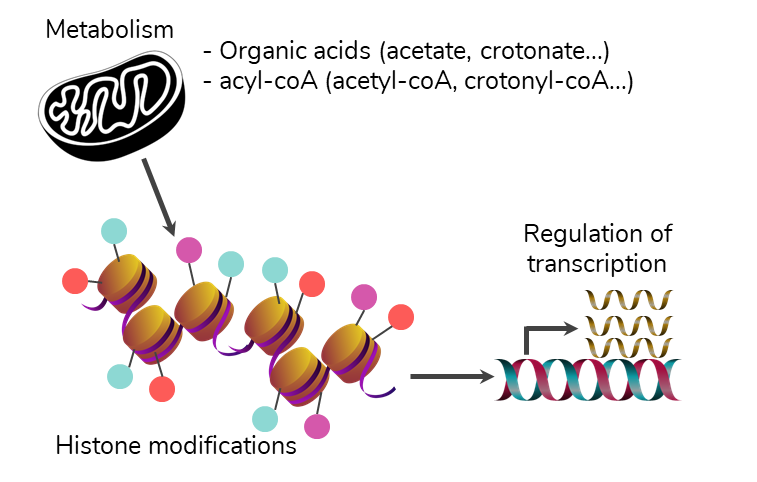
Now, we are building on our acquired expertise in integrating diverse omics data to study the effect of sparsely characterized histone PTMs on gene expression regulation. On the one hand, we are further studying histone lysine acylations in the context of mouse spermatogenesis. On the other hand, we are expanding such studies to biomedical contexts in which epigenetic landscapes are rewired. Huntington’s disease (HD) is a rare genetic neurodegenerative disease that primarily affects the striatum and leads to motor, cognitive and psychiatric symptoms. Early steps of HD pathogenesis involve altered histone acetylation in the striatum, the underlying mechanism of which is unclear. In collaboration with Karine Merienne (LCNA CNRS/University of Strasbourg), we wish to refine the picture of histone PTMs that are perturbed in HD, and thus lead to an aberrant transcription program in neurons.

Publications
- El Kennani S, Adrait S, Shaytan A, Khochbin S, Bruley C, Panchenko AR, Landsman D, Pflieger D*, Govin J*. MS_HistoneDB, a manually curated resource for proteomic analysis of human and mouse histones. Epigenetics & Chromatin, 2017, Jan 10;10:2. doi: 10.1186/s13072-016-0109-x.
- El Kennani S, Adrait A, Permiakova O, Hesse AM, Ialy-Radio C, Ferro M, Brun V, Cocquet J, Govin J*, Pflieger D*. Systematic quantitative analysis of H2A and H2B variants by targeted proteomics. Epigenetics & Chromatin, 2018, 2018 Jan 12;11(1):2. doi: 10.1186/s13072-017-0172-y.
- El Kennani S*, Crespo M, Govin J, Pflieger D*. Proteomic analysis of histone variants and their PTMs: strategies and pitfalls. Proteomes, 2018, 2018 Jun 21;6(3):29. doi: 10.3390/proteomes6030029.
- Marion Crespo, Annelaure Damont, Melina Blanco, Emmanuelle Lastrucci, Sara El Kennani, Côme Ialy-Radio, Laila El Khattabi, Samuel Terrier, Mathilde Louwagie, Sylvie Kieffer-Jaquinod, Anne-Marie Hesse, Christophe Bruley, Sophie Chantalat, Jérôme Govin, François Fenaille, Christophe Battail*, Julie Cocquet*, Delphine Pflieger*. Multi-omic analysis of gametogenesis reveals a novel signature at the promoters and distal enhancers of active genes. Nucleic Acids Res. 2020 May 7;48(8):4115-4138. doi: 10.1093/nar/gkaa163.
- Marion Crespo, Lacey J. Luense, Marie Arlotto, Jialei Hu, Jean Dorsey, Encar García-Oliver, Parisha P. Shah, Delphine Pflieger, Shelley L. Berger, Jerome Govin. Systematic genetic and proteomic screens during gametogenesis identify H2BK34 methylation as an evolutionary conserved meiotic mark. Epigenetics & Chromatin, 2020 Sep 15;13(1):35. doi: 10.1186/s13072-020-00349-5.
- Hseiky A, Crespo M, Kieffer-Jaquinod S, Fenaille F, Pflieger D*. Small Mass but Strong Information: Diagnostic Ions Provide Crucial Clues to Correctly Identify Histone Lysine Modifications. Proteomes. 2021 Apr 23;9(2):18. doi: 10.3390/proteomes9020018.
Fundings
- ANR funding on the project CHROMACYL (2022-2026): “Studying novel histone acylations in the regulation of gene expression”
- GRAL PhD fellowship to Hassan Hijazi (2021-2024): “HuntAcyl: Altered dynamics of acylations of histone H3 Lysine 27 (H3K27): a mechanism contributing to transcriptional dysregulation in Huntington’s disease?”
- UGA PhD fellowship to Marion Crespo (2016-2019)
- CEA PhD fellowship to Sara El Kennani (2015-2019)
Image credit: PublicDomainPictures de Pixabay